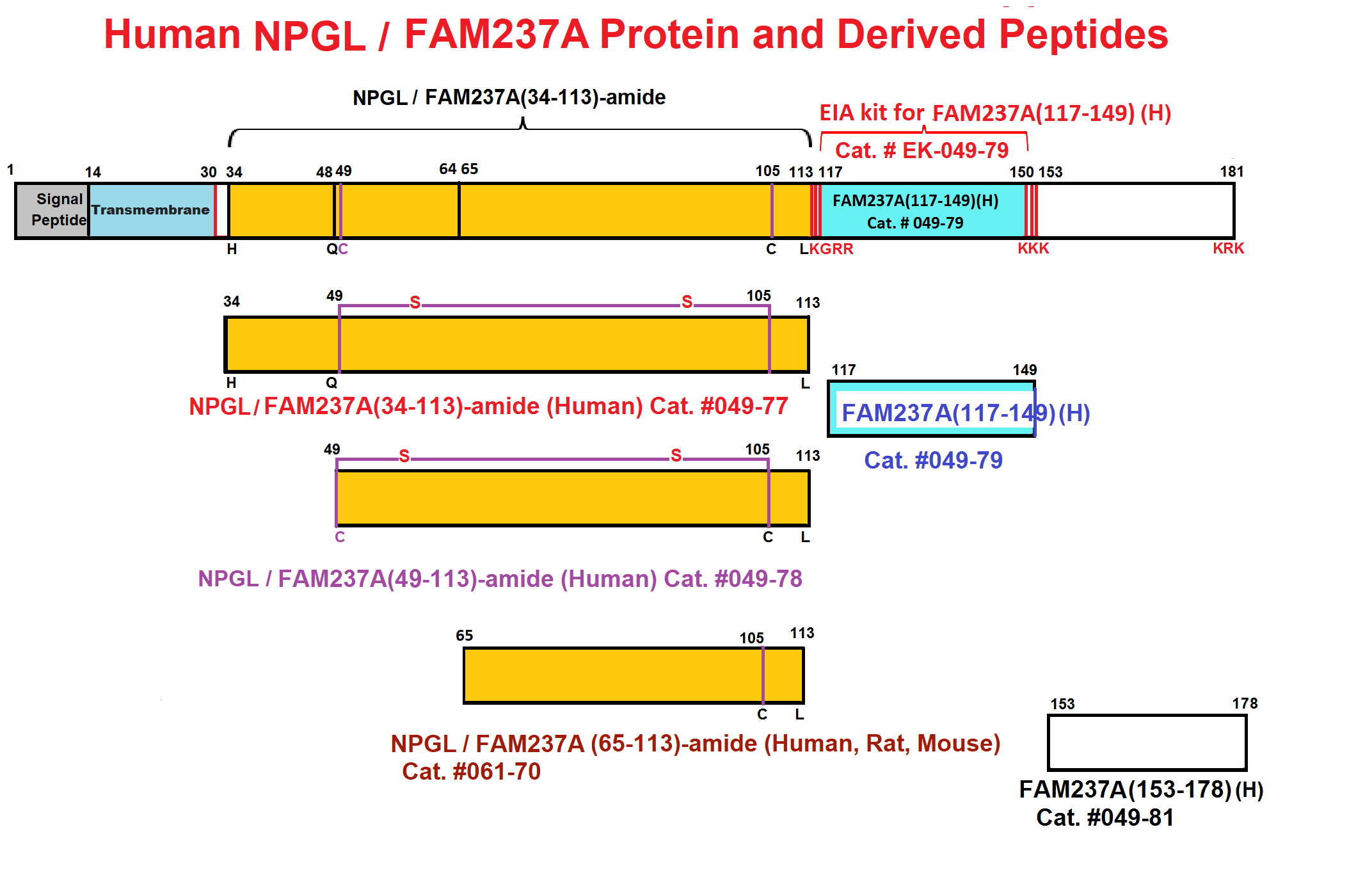


Microproteins (MPs) are a potentially rich source of uncharacterized metabolic regulators. Here, we use ribosome profiling (Ribo-seq) to curate 3,877 unannotated MP-encoding small ORFs (smORFs) in primary brown, white, and beige mouse adipocytes. Of these, we validated 85 MPs by proteomics, including 33 circulating MPs in mouse plasma. Analyses of MP-encoding mRNAs under different physiological conditions (high-fat diet) revealed that numerous MPs are regulated in adipose tissue in vivo and are co-expressed with established metabolic genes. Furthermore, Ribo-seq provided evidence for the translation of Gm8773, which encodes a secreted MP that is homologous to human and chicken FAM237B. Gm8773 is highly expressed in the arcuate nucleus of the hypothalamus, and intracerebroventricular administration of recombinant mFAM237B showed orexigenic activity in obese mice. Together, these data highlight the value of this adipocyte MP database in identifying MPs with roles in fundamental metabolic and physiological processes such as feeding.
Martinez TF, Lyons-Abbott S, Bookout AL, et al. Profiling mouse brown and white adipocytes to identify metabolically relevant small ORFs and functional microproteins. Cell Metabolism. 2023;35(1):166-183.e11.
Purpose: High-grade serous ovarian carcinoma (HGSOC) typically remains undiagnosed until advanced stages when peritoneal dissemination has already occurred. Here, we sought to identify HGSOC-specific alterations in DNA methylation and assess their potential to provide sensitive and specific detection of HGSOC at its earliest stages.Experimental design: Methylation EPIC genome-wide methylation analysis was performed on a discovery cohort comprising 23 HGSOC, 37 non-HGSOC malignant, and 36 histologically unremarkable gynecologic tissue samples. The resulting data were processed using selective bioinformatic criteria to identify regions of high-confidence HGSOC-specific differential methylation. Quantitative methylation-specific real-time PCR (qMSP) assays were then developed for 8 of the top-performing regions and analytically validated in a cohort of 90 tissue samples. Lastly, qMSP assays were used to assess and compare methylation in 30 laser-capture microdissected (LCM) fallopian tube epithelia samples obtained from cancer-free and serous tubal intraepithelial carcinoma (STIC) positive women.Results: Bioinformatic selection identified 91 regions of robust, HGSOC-specific hypermethylation, 23 of which exhibited an area under the receiver-operator curve (AUC) value ≥ 0.9 in the discovery cohort. Seven of 8 top-performing regions demonstrated AUC values between 0.838 and 0.968 when analytically validated by qMSP in a 90-patient cohort. A panel of the 3 top-performing genes (c17orf64, IRX2, and TUBB6) was able to perfectly discriminate HGSOC (AUC 1.0). Hypermethylation within these loci was found exclusively in LCM fallopian tube epithelia from women with STIC lesions, but not in cancer-free fallopian tubes.Conclusions: A panel of methylation biomarkers can be used to accurately identify HGSOC, even at precursor stages of the disease.
Pisanic TR, Cope LM, Lin SF, et al. Methylomic Analysis of Ovarian Cancers Identifies Tumor-Specific Alterations Readily Detectable in Early Precursor Lesions. Clin Cancer Res. 2018;24(24):6536-6547.
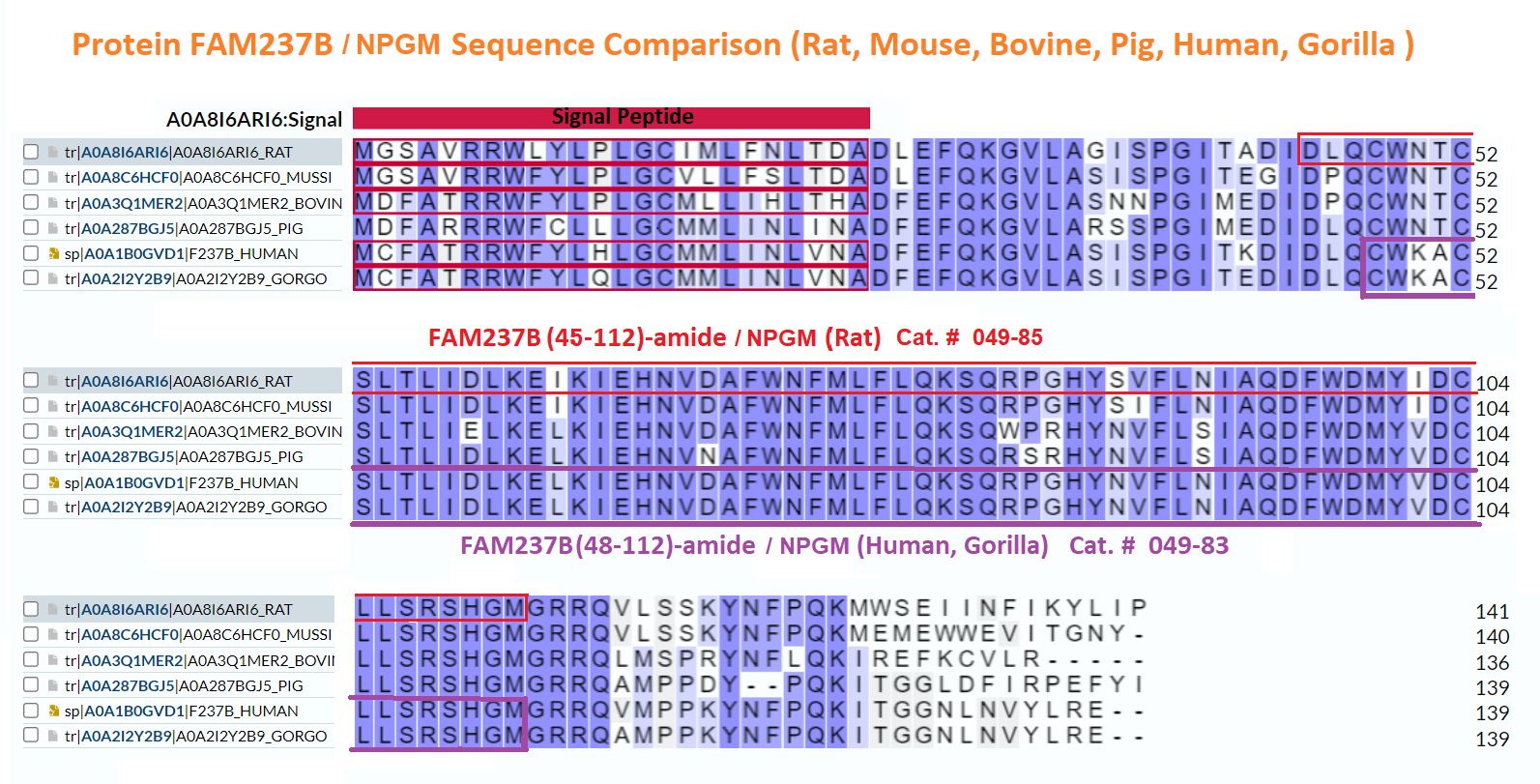
Abstract: The identification of novel peptide hormones by functional screening is challenging because posttranslational processing is frequently required to generate biologically active hormones from inactive precursors. We developed an approach for functional screening of novel potential hormones by expressing them in endocrine host cells competent for posttranslational processing. Candidate preprohormones were selected by bioinformatics analysis, and stable endocrine host cell lines were engineered to express the preprohormones. The production of mature hormones was demonstrated by including the preprohormones insulin and glucagon, which require the regulated secretory pathway for production of the active forms. As proof of concept, we screened a set of G-protein-coupled receptors (GPCRs) and identified protein FAM237A as a specific activator of GPR83, a GPCR implicated in central nervous system and regulatory T-cell function. We identified the active form of FAM237A as a C-terminally cleaved, amidated 9 kDa secreted protein. The related protein FAM237B, which is 64% homologous to FAM237A, demonstrated similar posttranslational modification and activation of GPR83, albeit with reduced potency. These results demonstrate that our approach is capable of identifying and characterizing novel hormones that require processing for activity.
Sallee NA, Lee E, Leffert A, et al. A Pilot Screen of a Novel Peptide Hormone Library Identified Candidate GPR83 Ligands. SLAS Discov. 2020;:2472555220934807.
| Catalog# | Product | Standard Size | Price |
|---|---|---|---|
| 049-89 | FAM237A (34-113)-Amide (Ryukyu Mouse) | 100 µg | $450 |
| 049-87 | FAM237A (34-113)-amide (Rat, House Mouse) | 100 μg | $450 |
| 049-79 | FAM237A / NPGL (117-149) (Human) | 100 µg | $356 |
| EK-049-79 | FAM237A / NPGL (117-149)(Human) – EIA kit | 96 wells | $548 |
| 049-81 | FAM237A / NPGL (153-178) (Human) | 100 µg | $296 |
| 049-78 | FAM237A / NPGL (49-113)-amide (Human) | 100 µg | $415 |
| 049-83 | FAM237B / NPGM (48-112)-amide (Human) | 100 µg | $450 |
| RK-049-83 | FAM237B / NPGM (48-112)-amide (Human) - RIA Kit | 125 tubes | $926 |
| H-049-83 | FAM237B / NPGM (48-112)-amide (Human)-- Antibody | 50 µl | $571 |
| G-049-83 | FAM237B / NPGM (48-112)-amide (Human)-Purified IgG Antibody | 200 µg | $698 |
Social Network Confirmation