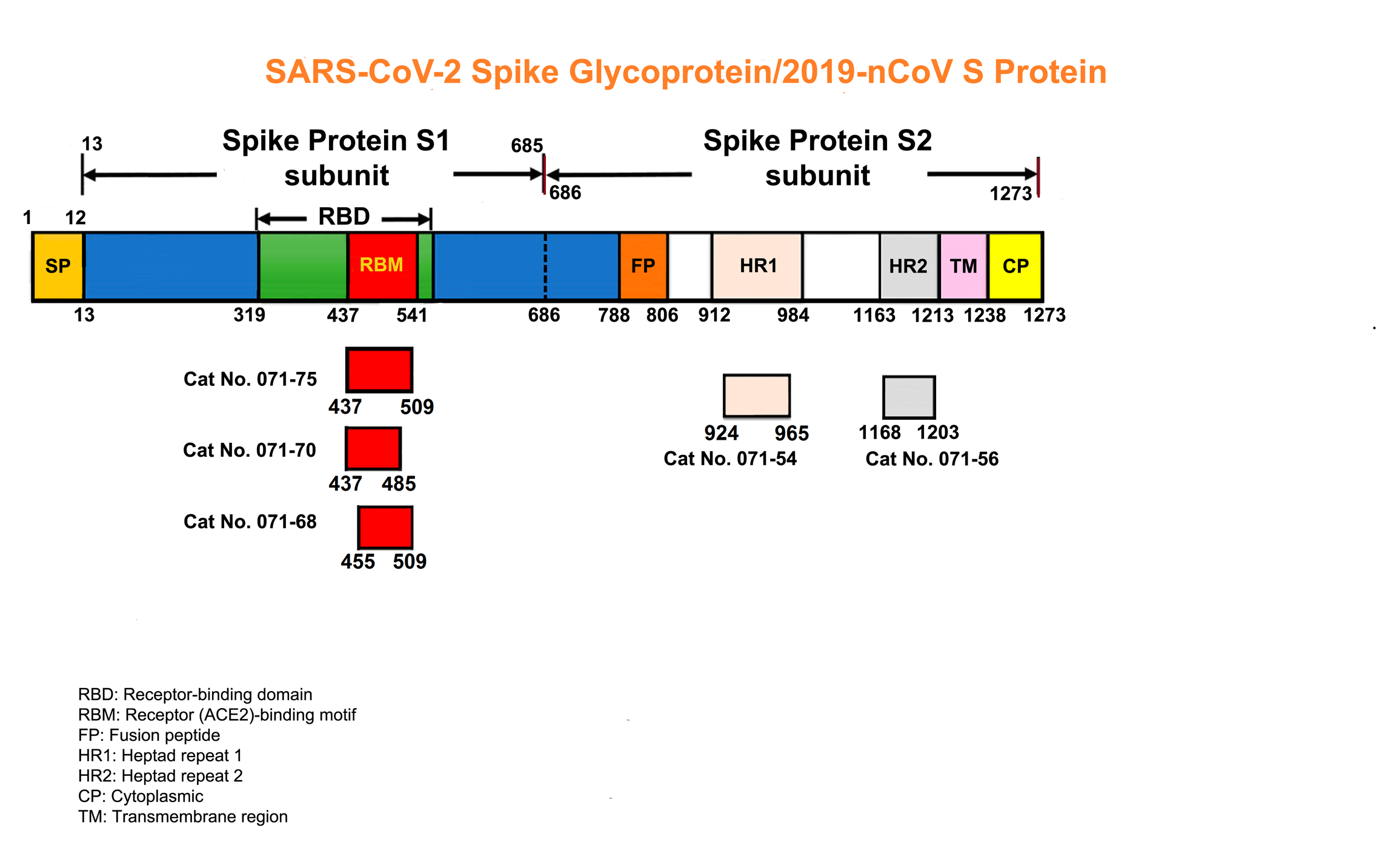
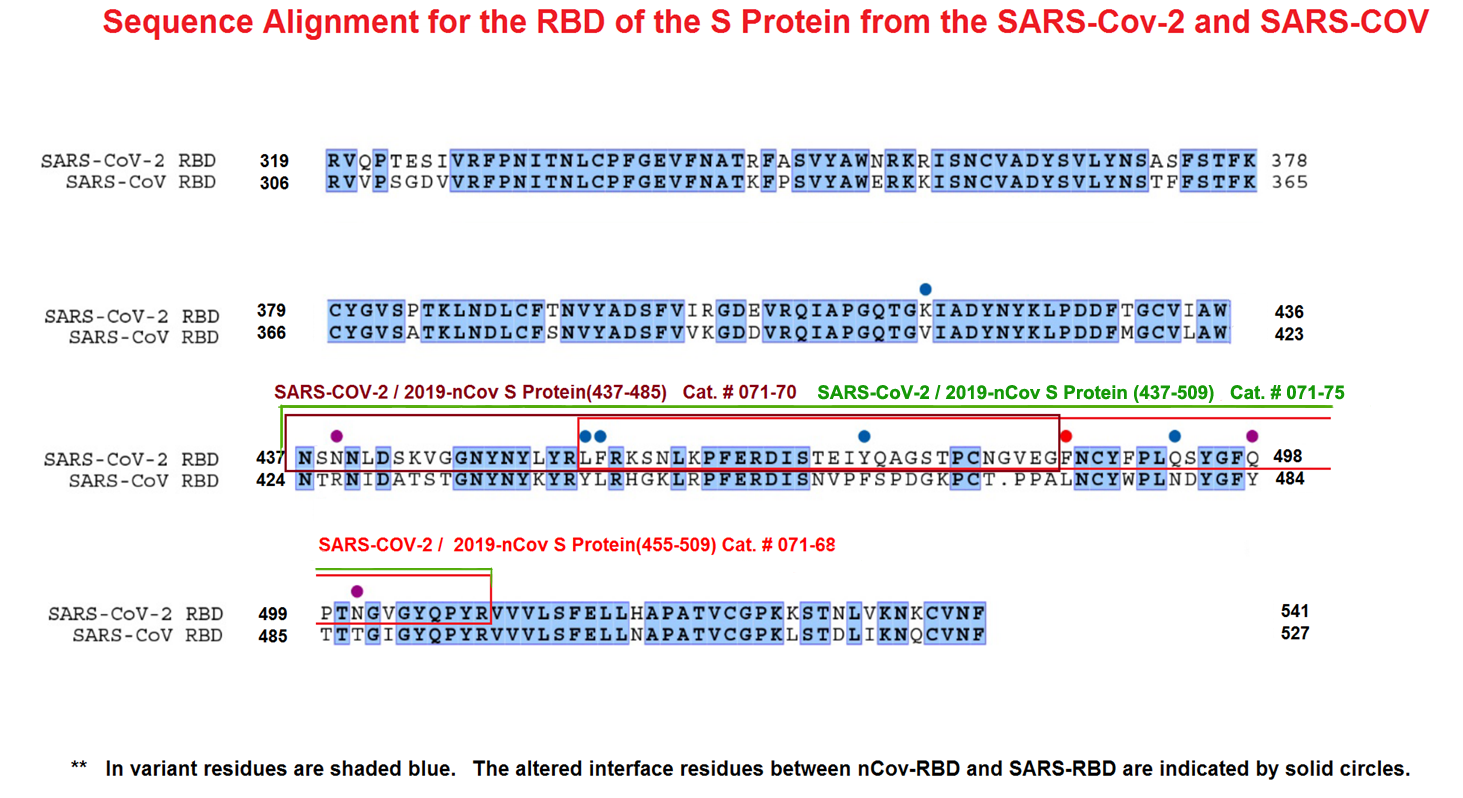
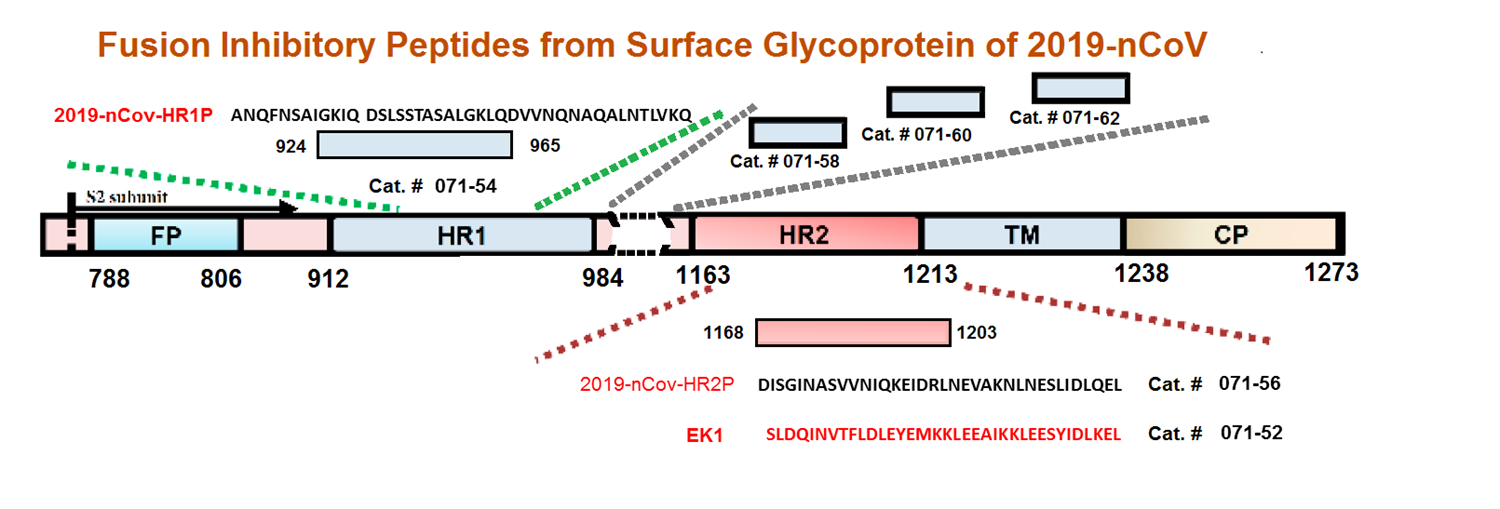


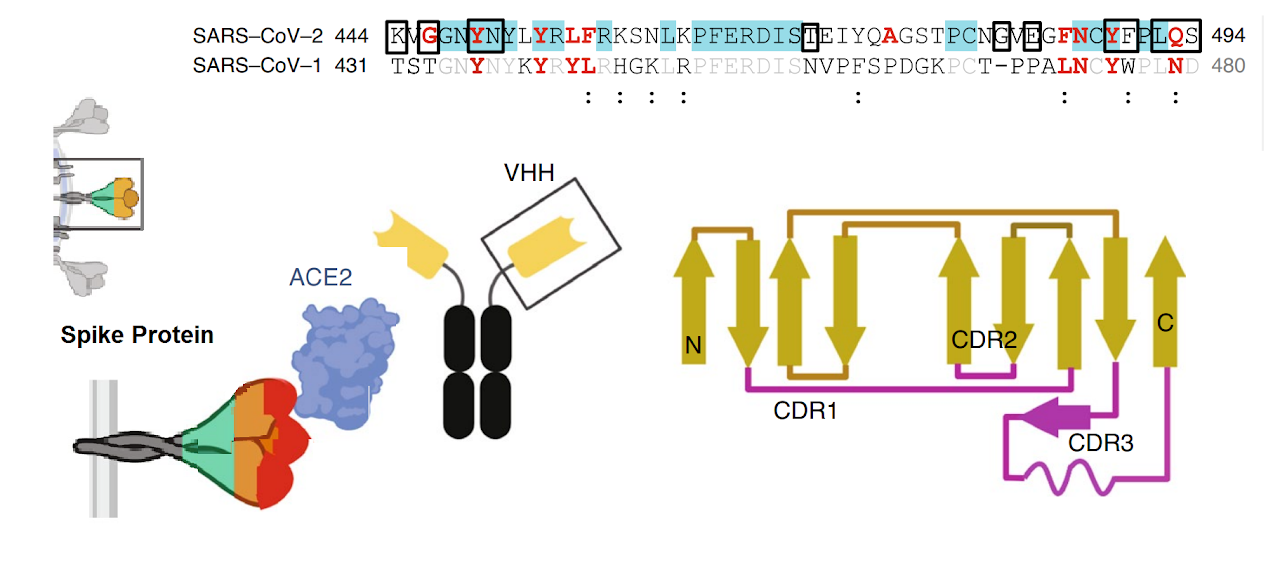
The spike protein of SARS-CoV-2 drives infection. The spike protein is composed of S1 and S2 subunits. S1 contains the RBD (highlighted in red). Using the RBD, the trimeric spoke molecule binds to ACE2 on human cells (a single ACE2 is shown in blue). RBD residues that are shared between SARS-CoV-2 and SARS-Cov-1 are highlited in cyan (SARS-CoV-2 top) or in light gray text (SARS-CoV-1, bottom). Resideus that contact ACE2 are highlited in red for each sequence. Other sequence differences are in black test, with conservative substitutions indivated by colons (:) underneath. Resideies in contact with the H11-H4 nanobody are boxed.
Figure from Jiangdong Huo et al. of Nat Struct Mol Biol . 2020 Jul 13. doi: 10.1038/s41594-020-0469-6
Background and purpose: Bradykinin (BK-(1-9)) is an endogenous nonapeptide involved in multiple physiological and pathological processes. A long-held belief is that peptide fragments of BK-(1-9) are biologically inactive. Here, we have tested the biological activities of BK-(1-9)’s two major peptide fragments in human and animal systems.
Experimental Approach: Levels of BK peptides in male Wistar rat plasma were quantified by mass spectrometry. NO release was quantified in human, mouse and rat cells, loaded with DAF-FM. We used aortic rings from adult male Wistar rats to test vascular reactivity. Changes in blood pressure and heart rate were measured in conscious adult male Wistar rats. Vascular permeability and nociception were measured in adult mice to evaluate potential pro-inflammatory effects.
Key results: Plasma levels of BK-(1-7) and BK-(1-5) in rats were increased following infusion of BK-(1-9). Both peptides induced NO production in all cell types tested. However, unlike BK-(1-9), NO production elicited by BK-(1-7) or BK-(1-5) was not inhibited by B1 or B2 receptor antagonists. BK-(1-7) and BK-(1-5) induced concentration-dependent vasorelaxation of aortic rings, without involvement B1 or B2 receptors. Intravenous or intra-arterial administration of BK-(1-7) or BK-(1-5) induced similar hypotensive response in vivo. Nociceptive responses of BK-(1-7) and BK-(1-5) were reduced when compared to BK-(1-9), and no increase of vascular permeability was observed for BK-(1-9) fragments.
Conclusions and implications: BK-(1-7) and BK-(1-5) are endogenous peptides present in plasma. BK-related peptide fragments show biological activity, not mediated by B1 or B2 receptors. These BK-fragments could constitute new, active components of the kallikrein-kinin system.
Souza-Silva IM, Paula CA de, Bolais-Ramos L, et al. Peptide fragments of bradykinin show unexpected biological activity not mediated by B1 or B2 receptors. British Journal of Pharmacology.

| Catalog# | Product | Standard Size | Price |
|---|---|---|---|
| 009-44 | BK-(1-5) / Bradykinin (1-5) | 1mg | $178 |
Social Network Confirmation