Catalog # |
Size |
Price |
|
|---|---|---|---|
| 034-75 | 100ug | $370 |
 )
)
|
Gly-Arg-Thr-Thr-Cys-Thr-Lys-Thr-Gln-Pro-Asn-Leu-Asp-Asn-Cys-Pro-Phe-His-Asp-Gln-Pro-His-Leu-Lys-Arg-Lys-Ala-Phe-Cys-Ser-Phe-Gln-Ile-Tyr-Ala-Val-Pro-Trp-Gln-Gly-Thr-Met-Thr-Leu-Ser-Lys-Ser-Thr-Cys-Gln-Asp-Ala
|
|
Cys99-Cys109 & Cys123-Cys143
|
| 5898.74 | |
|
| ≥ 94% |
|
| Exhibits correct molecular weight |
|
|
Up to 6 months in lyophilized form at 0-5ºC. For best results, rehydrate just before use. Aliquot before freezing to avoid repeated freeze-thaw cycles. |
|
| White powder |
|
| Each vial contains 100 μg of NET peptide. |
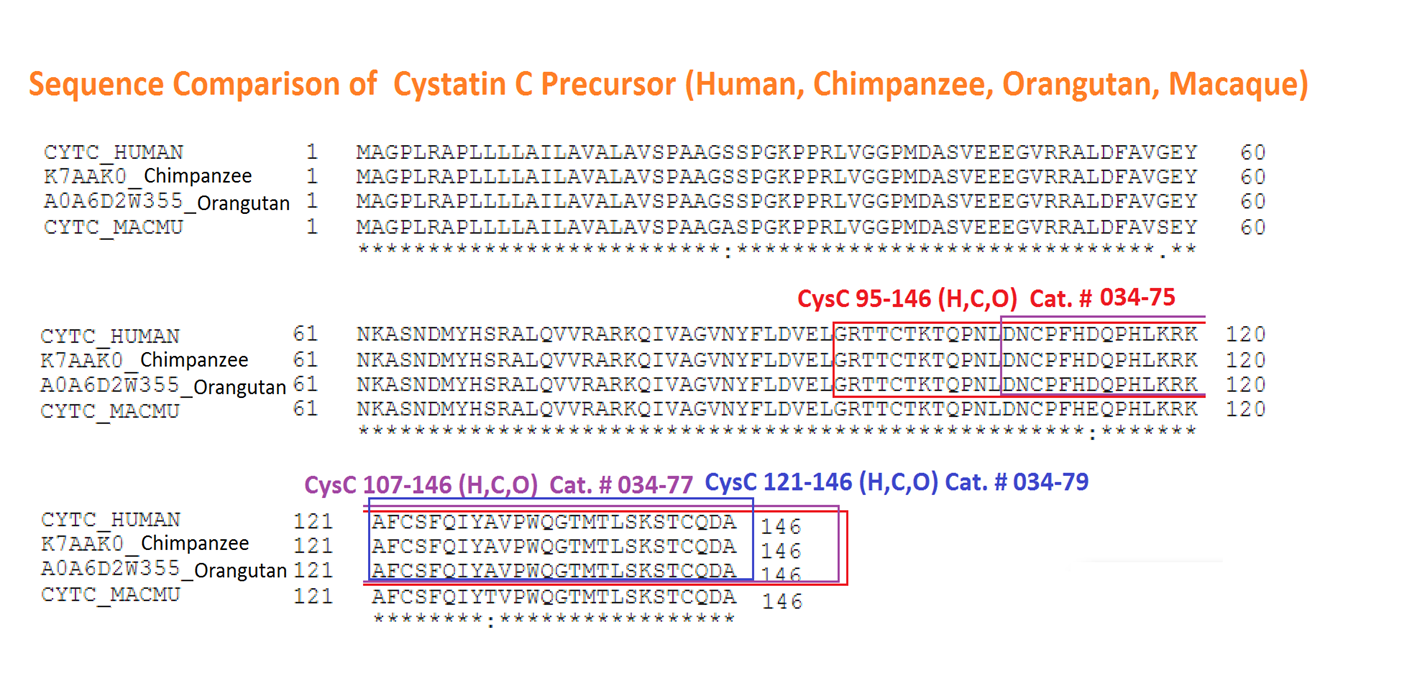
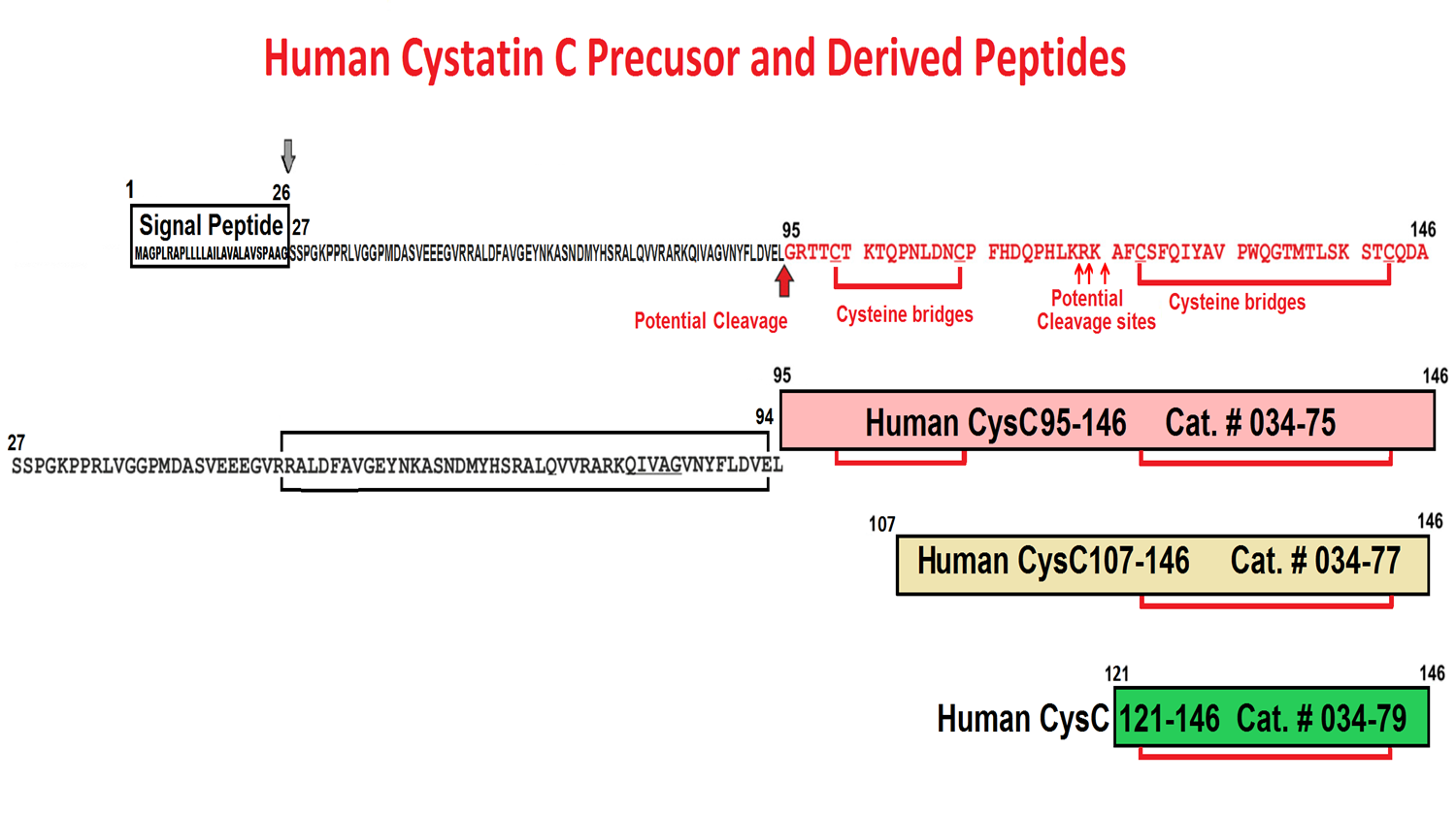
Abstract: GPR15 is a G protein-coupled receptor (GPCR) proposed to play a role in mucosal immunity that also serves as a major entry cofactor for HIV-2 and simian immunodeficiency virus (SIV). To discover novel endogenous GPR15 ligands, we screened a hemofiltrate (HF)-derived peptide library for inhibitors of GPR15-mediated SIV infection. Our approach identified a C-terminal fragment of cystatin C (CysC95-146) that specifically inhibits GPR15-dependent HIV-1, HIV-2, and SIV infection. In contrast, GPR15L, the chemokine ligand of GPR15, failed to inhibit virus infection. We found that cystatin C fragments preventing GPR15-mediated viral entry do not interfere with GPR15L signaling and are generated by proteases activated at sites of inflammation. The antiretroviral activity of CysC95-146 was confirmed in primary CD4+ T cells and is conserved in simian hosts of SIV infection. Thus, we identified a potent endogenous inhibitor of GPR15-mediated HIV and SIV infection that does not interfere with the physiological function of this GPCR.
Hayn M, Blötz A, Rodríguez A, et al. Natural cystatin C fragments inhibit GPR15-mediated HIV and SIV infection without interfering with GPR15L signaling.
Abstract: Human cystatin C (hCC) is a low molecular mass protein that belongs to the cystatin superfamily. It is an inhibitor of extracellular cysteine proteinases, present in all human body fluids. At physiological conditions, hCC is a monomer, but it has a tendency to dimerization. Naturally occurring hCC mutant, with leucine in position 68 substituted by glutamine (L68Q), is directly involved in the formation of amyloid deposits, independently of other proteins. This process is the primary cause of hereditary cerebral amyloid angiopathy, observed mainly in the Icelandic population. Oligomerization and fibrillization processes of hCC are not explained equally well, but it is proposed that domain swapping is involved in both of them. Research carried out on the fibrillization process led to new hypothesis about the existence of a steric zipper motif in amyloidogenic proteins. In the hCC sequence, there are 2 fragments which may play the role of a steric zipper: the loop L1 region and the C-terminal fragment. In this work, we focused on the first of these. Nine hexapeptides covering studied hCC fragment were synthesized, and their fibrillogenic potential was assessed using an array of biophysical methods. The obtained results showed that the studied hCC fragment has strong profibrillogenic propensities because it contains 2 fragments fulfilling the requirements for an effective steric zipper located next to each other, forming 1 super-steric zipper motif. This hCC fragment might therefore be responsible for the enhanced amyloidogenic properties of dimeric or partially unfolded hCC.
I?owska E, Sawicka J, Szyma?ska A. Synthesis and physicochemical studies of amyloidogenic hexapeptides derived from human cystatin C. J Pep Sci.
No References
Social Network Confirmation